Variational Autoencoder#
pimmslearn - INFO Median Imputation
Papermill script parameters:
# files and folders
folder_experiment: str = 'runs/example' # Datasplit folder with data for experiment
file_format: str = 'csv' # file format of create splits, default pickle (pkl)
fn_rawfile_metadata: str = 'data/dev_datasets/HeLa_6070/files_selected_metadata_N50.csv' # Metadata for samples
# model
sample_idx_position: int = 0 # position of index which is sample ID
model_key: str = 'Median' # model key (lower cased version will be used for file names)
model: str = 'Median' # model name
save_pred_real_na: bool = True # Save all predictions for real na
# metadata -> defaults for metadata extracted from machine data
meta_date_col: str = None # date column in meta data
meta_cat_col: str = None # category column in meta data
# Parameters
model = "Median"
fn_rawfile_metadata = "https://raw.githubusercontent.com/RasmussenLab/njab/HEAD/docs/tutorial/data/alzheimer/meta.csv"
folder_experiment = "runs/alzheimer_study"
model_key = "Median"
Some argument transformations
{'folder_experiment': 'runs/alzheimer_study',
'file_format': 'csv',
'fn_rawfile_metadata': 'https://raw.githubusercontent.com/RasmussenLab/njab/HEAD/docs/tutorial/data/alzheimer/meta.csv',
'sample_idx_position': 0,
'model_key': 'Median',
'model': 'Median',
'save_pred_real_na': True,
'meta_date_col': None,
'meta_cat_col': None}
{'data': Path('runs/alzheimer_study/data'),
'file_format': 'csv',
'fn_rawfile_metadata': 'https://raw.githubusercontent.com/RasmussenLab/njab/HEAD/docs/tutorial/data/alzheimer/meta.csv',
'folder_experiment': Path('runs/alzheimer_study'),
'meta_cat_col': None,
'meta_date_col': None,
'model': 'Median',
'model_key': 'Median',
'out_figures': Path('runs/alzheimer_study/figures'),
'out_folder': Path('runs/alzheimer_study'),
'out_metrics': Path('runs/alzheimer_study'),
'out_models': Path('runs/alzheimer_study'),
'out_preds': Path('runs/alzheimer_study/preds'),
'sample_idx_position': 0,
'save_pred_real_na': True}
Some naming conventions
Load data in long format#
pimmslearn.io.datasplits - INFO Loaded 'train_X' from file: runs/alzheimer_study/data/train_X.csv
pimmslearn.io.datasplits - INFO Loaded 'val_y' from file: runs/alzheimer_study/data/val_y.csv
pimmslearn.io.datasplits - INFO Loaded 'test_y' from file: runs/alzheimer_study/data/test_y.csv
data is loaded in long format
Sample ID protein groups
Sample_041 J3KRI5;J3QKW5;J3QLE6;P55286;X6R3Y6 16.305
Sample_141 P78417 15.433
Sample_078 Q96ID5 15.943
Sample_095 I3L3E6;Q658N2 15.668
Sample_059 P13671 19.715
Name: intensity, dtype: float64
Infer index names from long format
pimmslearn - INFO sample_id = 'Sample ID', single feature: index_column = 'protein groups'
load meta data for splits
| _collection site | _age at CSF collection | _gender | _t-tau [ng/L] | _p-tau [ng/L] | _Abeta-42 [ng/L] | _Abeta-40 [ng/L] | _Abeta-42/Abeta-40 ratio | _primary biochemical AD classification | _clinical AD diagnosis | _MMSE score | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample ID | |||||||||||
| Sample_000 | Sweden | 71.000 | f | 703.000 | 85.000 | 562.000 | NaN | NaN | biochemical control | NaN | NaN |
| Sample_001 | Sweden | 77.000 | m | 518.000 | 91.000 | 334.000 | NaN | NaN | biochemical AD | NaN | NaN |
| Sample_002 | Sweden | 75.000 | m | 974.000 | 87.000 | 515.000 | NaN | NaN | biochemical AD | NaN | NaN |
| Sample_003 | Sweden | 72.000 | f | 950.000 | 109.000 | 394.000 | NaN | NaN | biochemical AD | NaN | NaN |
| Sample_004 | Sweden | 63.000 | f | 873.000 | 88.000 | 234.000 | NaN | NaN | biochemical AD | NaN | NaN |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| Sample_205 | Berlin | 69.000 | f | 1,945.000 | NaN | 699.000 | 12,140.000 | 0.058 | biochemical AD | AD | 17.000 |
| Sample_206 | Berlin | 73.000 | m | 299.000 | NaN | 1,420.000 | 16,571.000 | 0.086 | biochemical control | non-AD | 28.000 |
| Sample_207 | Berlin | 71.000 | f | 262.000 | NaN | 639.000 | 9,663.000 | 0.066 | biochemical control | non-AD | 28.000 |
| Sample_208 | Berlin | 83.000 | m | 289.000 | NaN | 1,436.000 | 11,285.000 | 0.127 | biochemical control | non-AD | 24.000 |
| Sample_209 | Berlin | 63.000 | f | 591.000 | NaN | 1,299.000 | 11,232.000 | 0.116 | biochemical control | non-AD | 29.000 |
210 rows × 11 columns
Initialize Comparison#
replicates idea for truely missing values: Define truth as by using n=3 replicates to impute each sample
real test data:
Not used for predictions or early stopping.
[x] add some additional NAs based on distribution of data
protein groups
A0A024QZX5;A0A087X1N8;P35237 197
A0A024R0T9;K7ER74;P02655 208
A0A024R3W6;A0A024R412;O60462;O60462-2;O60462-3;O60462-4;O60462-5;Q7LBX6;X5D2Q8 185
A0A024R644;A0A0A0MRU5;A0A1B0GWI2;O75503 208
A0A075B6H7 97
Name: freq, dtype: int64
Produce some addional fake samples#
The validation fake NA is used to by all models to evaluate training performance.
| observed | ||
|---|---|---|
| Sample ID | protein groups | |
| Sample_158 | Q9UN70;Q9UN70-2 | 14.630 |
| Sample_050 | Q9Y287 | 15.755 |
| Sample_107 | Q8N475;Q8N475-2 | 15.029 |
| Sample_199 | P06307 | 19.376 |
| Sample_067 | Q5VUB5 | 15.309 |
| ... | ... | ... |
| Sample_111 | F6SYF8;Q9UBP4 | 22.822 |
| Sample_002 | A0A0A0MT36 | 18.165 |
| Sample_049 | Q8WY21;Q8WY21-2;Q8WY21-3;Q8WY21-4 | 15.525 |
| Sample_182 | Q8NFT8 | 14.379 |
| Sample_123 | Q16853;Q16853-2 | 14.504 |
12600 rows × 1 columns
| observed | |
|---|---|
| count | 12,600.000 |
| mean | 16.339 |
| std | 2.741 |
| min | 7.209 |
| 25% | 14.412 |
| 50% | 15.935 |
| 75% | 17.910 |
| max | 30.140 |
Data in wide format#
Autoencoder need data in wide format
| protein groups | A0A024QZX5;A0A087X1N8;P35237 | A0A024R0T9;K7ER74;P02655 | A0A024R3W6;A0A024R412;O60462;O60462-2;O60462-3;O60462-4;O60462-5;Q7LBX6;X5D2Q8 | A0A024R644;A0A0A0MRU5;A0A1B0GWI2;O75503 | A0A075B6H7 | A0A075B6H9 | A0A075B6I0 | A0A075B6I1 | A0A075B6I6 | A0A075B6I9 | ... | Q9Y653;Q9Y653-2;Q9Y653-3 | Q9Y696 | Q9Y6C2 | Q9Y6N6 | Q9Y6N7;Q9Y6N7-2;Q9Y6N7-4 | Q9Y6R7 | Q9Y6X5 | Q9Y6Y8;Q9Y6Y8-2 | Q9Y6Y9 | S4R3U6 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample ID | |||||||||||||||||||||
| Sample_000 | 15.912 | 16.852 | 15.570 | 16.481 | 17.301 | 20.246 | 16.764 | 17.584 | 16.988 | 20.054 | ... | 16.012 | 15.178 | NaN | 15.050 | 16.842 | NaN | NaN | 19.563 | NaN | 12.805 |
| Sample_001 | NaN | 16.874 | 15.519 | 16.387 | NaN | 19.941 | 18.786 | 17.144 | NaN | 19.067 | ... | 15.528 | 15.576 | NaN | 14.833 | 16.597 | 20.299 | 15.556 | 19.386 | 13.970 | 12.442 |
| Sample_002 | 16.111 | NaN | 15.935 | 16.416 | 18.175 | 19.251 | 16.832 | 15.671 | 17.012 | 18.569 | ... | 15.229 | 14.728 | 13.757 | 15.118 | 17.440 | 19.598 | 15.735 | 20.447 | 12.636 | 12.505 |
| Sample_003 | 16.107 | 17.032 | 15.802 | 16.979 | 15.963 | 19.628 | 17.852 | 18.877 | 14.182 | 18.985 | ... | 15.495 | 14.590 | 14.682 | 15.140 | 17.356 | 19.429 | NaN | 20.216 | NaN | 12.445 |
| Sample_004 | 15.603 | 15.331 | 15.375 | 16.679 | NaN | 20.450 | 18.682 | 17.081 | 14.140 | 19.686 | ... | 14.757 | NaN | NaN | 15.256 | 17.075 | 19.582 | 15.328 | NaN | 13.145 | NaN |
5 rows × 1421 columns
Add interpolation performance#
| observed | Median | ||
|---|---|---|---|
| Sample ID | protein groups | ||
| Sample_158 | Q9UN70;Q9UN70-2 | 14.630 | 15.752 |
| Sample_050 | Q9Y287 | 15.755 | 17.221 |
| Sample_107 | Q8N475;Q8N475-2 | 15.029 | 14.846 |
| Sample_199 | P06307 | 19.376 | 18.973 |
| Sample_067 | Q5VUB5 | 15.309 | 14.726 |
| ... | ... | ... | ... |
| Sample_111 | F6SYF8;Q9UBP4 | 22.822 | 22.918 |
| Sample_002 | A0A0A0MT36 | 18.165 | 15.877 |
| Sample_049 | Q8WY21;Q8WY21-2;Q8WY21-3;Q8WY21-4 | 15.525 | 16.278 |
| Sample_182 | Q8NFT8 | 14.379 | 13.995 |
| Sample_123 | Q16853;Q16853-2 | 14.504 | 14.849 |
12600 rows × 2 columns
| Median | ||
|---|---|---|
| Sample ID | protein groups | |
| Sample_000 | A0A075B6J9 | 16.691 |
| A0A075B6Q5 | 16.503 | |
| A0A075B6R2 | 17.090 | |
| A0A075B6S5 | 16.203 | |
| A0A087WSY4 | 15.732 | |
| ... | ... | ... |
| Sample_209 | Q9P1W8;Q9P1W8-2;Q9P1W8-4 | 16.019 |
| Q9UI40;Q9UI40-2 | 16.744 | |
| Q9UIW2 | 15.057 | |
| Q9UMX0;Q9UMX0-2;Q9UMX0-4 | 13.739 | |
| Q9UP79 | 15.525 |
46401 rows × 1 columns
Plots#
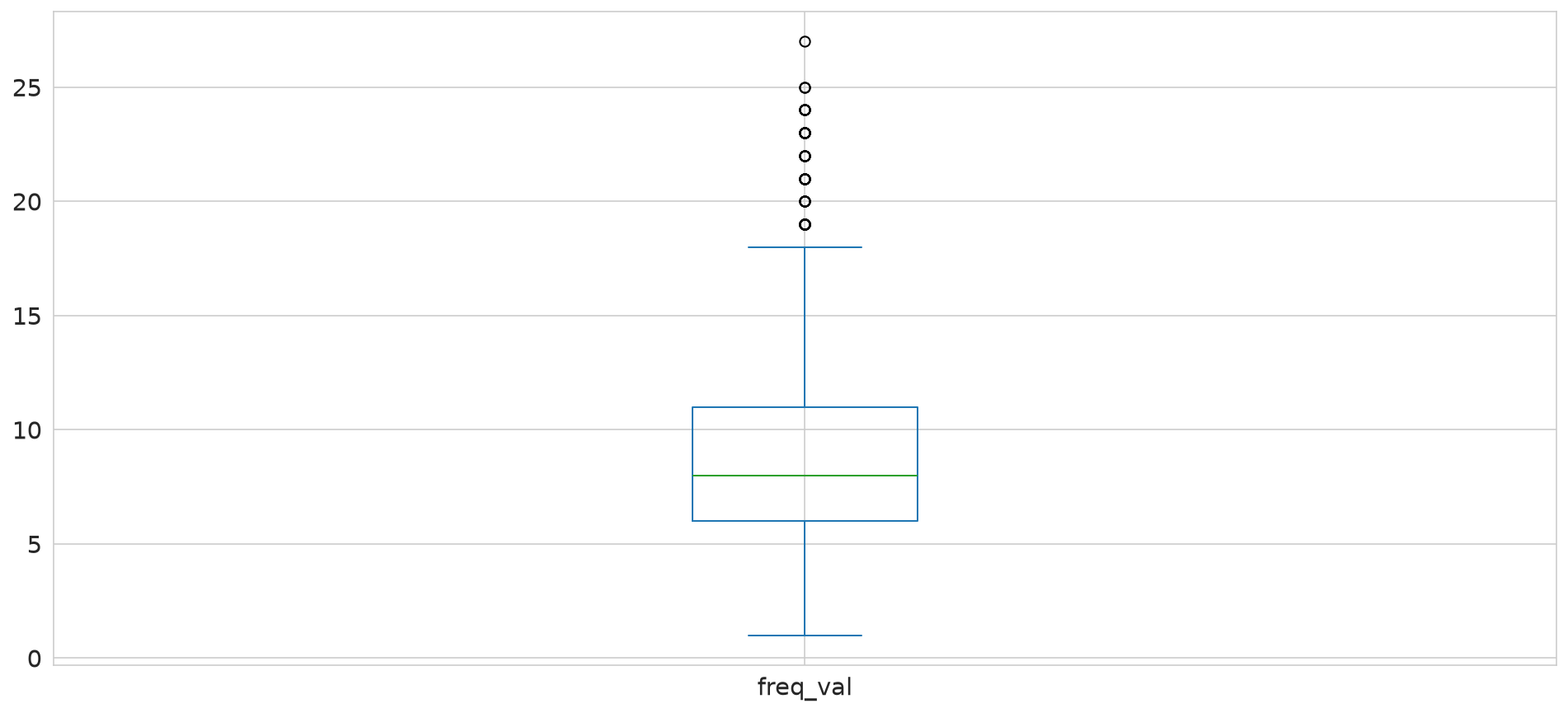
freq_val
1 12
2 18
3 50
4 82
5 108
Name: count, dtype: int64
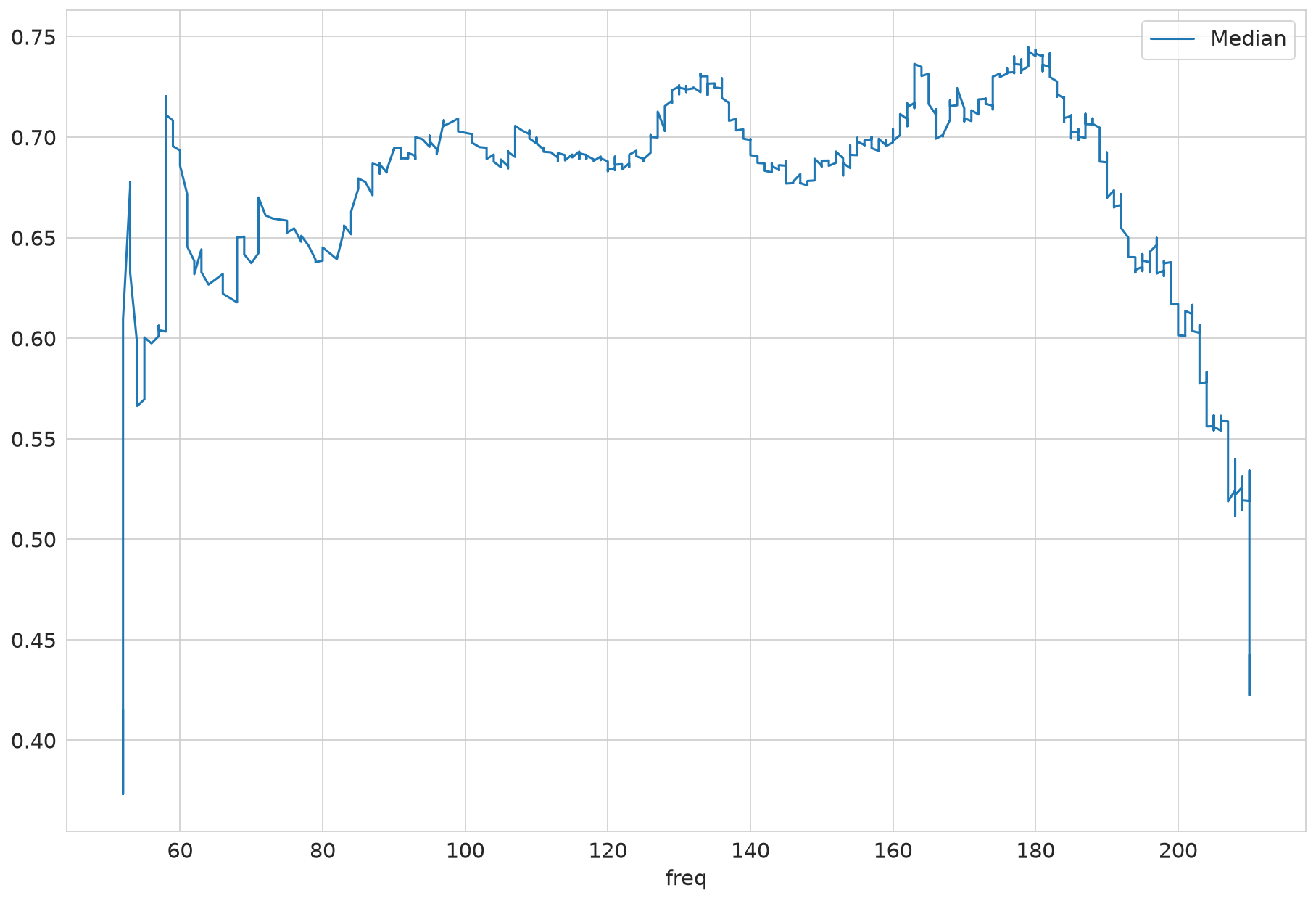
| Median | ||
|---|---|---|
| mean | count | |
| protein groups | ||
| A0A024QZX5;A0A087X1N8;P35237 | 0.270 | 7 |
| A0A024R0T9;K7ER74;P02655 | 1.713 | 4 |
| A0A024R3W6;A0A024R412;O60462;O60462-2;O60462-3;O60462-4;O60462-5;Q7LBX6;X5D2Q8 | 0.361 | 9 |
| A0A024R644;A0A0A0MRU5;A0A1B0GWI2;O75503 | 0.451 | 6 |
| A0A075B6H7 | 0.773 | 6 |
| ... | ... | ... |
| Q9Y6R7 | 0.489 | 10 |
| Q9Y6X5 | 0.538 | 7 |
| Q9Y6Y8;Q9Y6Y8-2 | 0.454 | 9 |
| Q9Y6Y9 | 0.812 | 15 |
| S4R3U6 | 0.518 | 24 |
1419 rows × 2 columns
| Median | ||
|---|---|---|
| Sample ID | protein groups | |
| Sample_158 | Q9UN70;Q9UN70-2 | 1.122 |
| Sample_050 | Q9Y287 | 1.466 |
| Sample_107 | Q8N475;Q8N475-2 | -0.183 |
| Sample_199 | P06307 | -0.403 |
| Sample_067 | Q5VUB5 | -0.583 |
| ... | ... | ... |
| Sample_111 | F6SYF8;Q9UBP4 | 0.096 |
| Sample_002 | A0A0A0MT36 | -2.288 |
| Sample_049 | Q8WY21;Q8WY21-2;Q8WY21-3;Q8WY21-4 | 0.753 |
| Sample_182 | Q8NFT8 | -0.383 |
| Sample_123 | Q16853;Q16853-2 | 0.345 |
12600 rows × 1 columns
Comparisons#
Validation data#
The fake NA for the validation step are real test data (not used for training nor early stopping)
Selected as truth to compare to: observed
{'Median': {'MSE': 0.766059536868567,
'MAE': 0.5984580373149887,
'N': 12600,
'prop': 1.0}}
Test Datasplit#
Fake NAs : Artificially created NAs. Some data was sampled and set explicitly to misssing before it was fed to the model for reconstruction.
Selected as truth to compare to: observed
{'Median': {'MSE': 0.7764184180515227,
'MAE': 0.6020439727512928,
'N': 12600,
'prop': 1.0}}
The fake NA for the validation step are real test data (not used for training nor early stopping)
Save all metrics as json#
{ 'test_fake_na': { 'Median': { 'MAE': 0.6020439727512928,
'MSE': 0.7764184180515227,
'N': 12600,
'prop': 1.0}},
'valid_fake_na': { 'Median': { 'MAE': 0.5984580373149887,
'MSE': 0.766059536868567,
'N': 12600,
'prop': 1.0}}}
| subset | valid_fake_na | test_fake_na | |
|---|---|---|---|
| model | metric_name | ||
| Median | MSE | 0.766 | 0.776 |
| MAE | 0.598 | 0.602 | |
| N | 12,600.000 | 12,600.000 | |
| prop | 1.000 | 1.000 |
Save predictions#
Config#
{}
{'M': 1421,
'data': Path('runs/alzheimer_study/data'),
'file_format': 'csv',
'fn_rawfile_metadata': 'https://raw.githubusercontent.com/RasmussenLab/njab/HEAD/docs/tutorial/data/alzheimer/meta.csv',
'folder_experiment': Path('runs/alzheimer_study'),
'meta_cat_col': None,
'meta_date_col': None,
'model': 'Median',
'model_key': 'Median',
'n_params': 1421,
'out_figures': Path('runs/alzheimer_study/figures'),
'out_folder': Path('runs/alzheimer_study'),
'out_metrics': Path('runs/alzheimer_study'),
'out_models': Path('runs/alzheimer_study'),
'out_preds': Path('runs/alzheimer_study/preds'),
'pred_test_Median': 'runs/alzheimer_study/preds/pred_test_Median.csv',
'pred_val_Median': 'runs/alzheimer_study/preds/pred_val_Median.csv',
'sample_idx_position': 0,
'save_pred_real_na': True}